Tutorial
In the following we will demonstrate the epmatools package. It is currently capable of doing just few impressive things:
[1]:
from epmatools import *
Basic usage
We should start with importing some data as oxides in wt%. You can use Oxides methods from_excel() or from_clipboard(), but for now we will use some example data provided:
[2]:
d = Oxides.from_examples('minerals')
d
[2]:
| SiO2 | Al2O3 | MgO | FeO | MnO | CaO | K2O | Na2O | TiO2 | Cr2O3 | ZnO | P2O5 | Y2O3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 37.218 | 20.349 | 13.784 | 13.140 | 0.000 | 0.013 | 8.998 | 0.404 | 1.010 | 0.022 | 0.020 | 0.005 | 0.007 |
| 1 | 37.363 | 20.037 | 14.106 | 12.539 | 0.000 | 0.013 | 8.946 | 0.461 | 1.206 | 0.020 | 0.042 | 0.000 | 0.000 |
| 2 | 23.748 | 22.152 | 9.611 | 31.549 | 0.066 | 0.028 | 0.023 | 0.035 | 0.047 | 0.000 | 0.017 | 0.037 | 0.000 |
| 3 | 46.986 | 39.183 | 0.151 | 1.264 | 0.033 | 0.410 | 3.409 | 4.883 | 0.083 | 0.000 | 0.000 | 0.006 | 0.000 |
| 4 | 48.389 | 32.414 | 8.227 | 7.713 | 0.060 | 0.027 | 0.000 | 0.924 | 0.005 | 0.009 | 0.000 | 0.005 | 0.000 |
| 5 | 48.870 | 32.696 | 8.390 | 7.482 | 0.068 | 0.049 | 0.000 | 0.845 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 6 | 61.839 | 24.661 | 0.000 | 0.484 | 0.015 | 5.738 | 0.032 | 7.917 | 0.000 | 0.010 | 0.019 | 0.025 | 0.000 |
| 7 | 37.816 | 21.184 | 4.189 | 35.118 | 1.001 | 1.435 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 | 0.000 |
| 8 | 37.584 | 21.311 | 4.651 | 33.895 | 0.999 | 1.454 | 0.000 | 0.035 | 0.013 | 0.034 | 0.000 | 0.030 | 0.107 |
| 9 | 60.657 | 25.087 | 0.001 | 0.000 | 0.000 | 5.849 | 0.106 | 7.940 | 0.000 | 0.000 | 0.003 | 0.113 | 0.000 |
| 10 | 61.338 | 25.025 | 0.015 | 0.030 | 0.003 | 5.801 | 0.079 | 7.997 | 0.047 | 0.000 | 0.057 | 0.064 | 0.000 |
| 11 | 28.057 | 53.950 | 2.040 | 11.888 | 0.186 | 0.036 | 0.000 | 0.000 | 0.585 | 0.063 | 1.024 | 0.000 | 0.000 |
| 12 | 27.565 | 53.885 | 1.972 | 11.828 | 0.175 | 0.000 | 0.000 | 0.000 | 0.607 | 0.003 | 1.035 | 0.000 | 0.000 |
| 13 | 27.091 | 53.841 | 2.099 | 11.875 | 0.180 | 0.031 | 0.000 | 0.000 | 0.554 | 0.046 | 1.116 | 0.000 | 0.000 |
| 14 | 46.078 | 35.793 | 0.652 | 0.784 | 0.000 | 0.002 | 9.333 | 1.182 | 0.697 | 0.081 | 0.000 | 0.000 | 0.000 |
| 15 | 45.487 | 35.669 | 0.803 | 0.895 | 0.015 | 0.000 | 9.394 | 1.171 | 0.643 | 0.033 | 0.015 | 0.000 | 0.034 |
Oxides automatically recognize valid column names for oxides, which are shown by default but other columns are also available. You can check them using property others
[3]:
d.others
[3]:
| Total | Comment | |
|---|---|---|
| 0 | 95.467 | bt-01 |
| 1 | 95.173 | bt-02 |
| 2 | 87.475 | chl-04 |
| 3 | 96.458 | pa-05 |
| 4 | 97.784 | cd-06 |
| 5 | 98.418 | cd-07 |
| 6 | 100.740 | pl-08 |
| 7 | 100.752 | g-09 |
| 8 | 100.124 | g-10 |
| 9 | 99.756 | pl-22 |
| 10 | 100.456 | pl-23 |
| 11 | 97.834 | st-33 |
| 12 | 97.070 | st-34 |
| 13 | 96.844 | st-35 |
| 14 | 94.612 | ms-49 |
| 15 | 94.159 | ms-50 |
Those columns could be set as index allowing to select and seach particular data
[4]:
d = d.set_index('Comment')
d
[4]:
| SiO2 | Al2O3 | MgO | FeO | MnO | CaO | K2O | Na2O | TiO2 | Cr2O3 | ZnO | P2O5 | Y2O3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| bt-01 | 37.218 | 20.349 | 13.784 | 13.140 | 0.000 | 0.013 | 8.998 | 0.404 | 1.010 | 0.022 | 0.020 | 0.005 | 0.007 |
| bt-02 | 37.363 | 20.037 | 14.106 | 12.539 | 0.000 | 0.013 | 8.946 | 0.461 | 1.206 | 0.020 | 0.042 | 0.000 | 0.000 |
| chl-04 | 23.748 | 22.152 | 9.611 | 31.549 | 0.066 | 0.028 | 0.023 | 0.035 | 0.047 | 0.000 | 0.017 | 0.037 | 0.000 |
| pa-05 | 46.986 | 39.183 | 0.151 | 1.264 | 0.033 | 0.410 | 3.409 | 4.883 | 0.083 | 0.000 | 0.000 | 0.006 | 0.000 |
| cd-06 | 48.389 | 32.414 | 8.227 | 7.713 | 0.060 | 0.027 | 0.000 | 0.924 | 0.005 | 0.009 | 0.000 | 0.005 | 0.000 |
| cd-07 | 48.870 | 32.696 | 8.390 | 7.482 | 0.068 | 0.049 | 0.000 | 0.845 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| pl-08 | 61.839 | 24.661 | 0.000 | 0.484 | 0.015 | 5.738 | 0.032 | 7.917 | 0.000 | 0.010 | 0.019 | 0.025 | 0.000 |
| g-09 | 37.816 | 21.184 | 4.189 | 35.118 | 1.001 | 1.435 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 | 0.000 |
| g-10 | 37.584 | 21.311 | 4.651 | 33.895 | 0.999 | 1.454 | 0.000 | 0.035 | 0.013 | 0.034 | 0.000 | 0.030 | 0.107 |
| pl-22 | 60.657 | 25.087 | 0.001 | 0.000 | 0.000 | 5.849 | 0.106 | 7.940 | 0.000 | 0.000 | 0.003 | 0.113 | 0.000 |
| pl-23 | 61.338 | 25.025 | 0.015 | 0.030 | 0.003 | 5.801 | 0.079 | 7.997 | 0.047 | 0.000 | 0.057 | 0.064 | 0.000 |
| st-33 | 28.057 | 53.950 | 2.040 | 11.888 | 0.186 | 0.036 | 0.000 | 0.000 | 0.585 | 0.063 | 1.024 | 0.000 | 0.000 |
| st-34 | 27.565 | 53.885 | 1.972 | 11.828 | 0.175 | 0.000 | 0.000 | 0.000 | 0.607 | 0.003 | 1.035 | 0.000 | 0.000 |
| st-35 | 27.091 | 53.841 | 2.099 | 11.875 | 0.180 | 0.031 | 0.000 | 0.000 | 0.554 | 0.046 | 1.116 | 0.000 | 0.000 |
| ms-49 | 46.078 | 35.793 | 0.652 | 0.784 | 0.000 | 0.002 | 9.333 | 1.182 | 0.697 | 0.081 | 0.000 | 0.000 | 0.000 |
| ms-50 | 45.487 | 35.669 | 0.803 | 0.895 | 0.015 | 0.000 | 9.394 | 1.171 | 0.643 | 0.033 | 0.015 | 0.000 | 0.034 |
To select just subset of data, we can use search() method. Using string argument, you can search for text in index, using numeric, you can select particular analysis. Note, that index could be modified by reset_index() and set_index() methods.
[5]:
g = d.search("g")
g
[5]:
| SiO2 | Al2O3 | MgO | FeO | MnO | CaO | K2O | Na2O | TiO2 | Cr2O3 | ZnO | P2O5 | Y2O3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g-09 | 37.816 | 21.184 | 4.189 | 35.118 | 1.001 | 1.435 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 | 0.000 |
| g-10 | 37.584 | 21.311 | 4.651 | 33.895 | 0.999 | 1.454 | 0.000 | 0.035 | 0.013 | 0.034 | 0.000 | 0.030 | 0.107 |
Analyses could be converted to cations p.f.u, either providing number of oxygens
[6]:
g.cations(noxy=12)
[6]:
| Si{4+} | Al{3+} | Mg{2+} | Fe{2+} | Mn{2+} | Ca{2+} | K{+} | Na{+} | Ti{4+} | Cr{3+} | Zn{2+} | P{5+} | Y{3+} | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g-09 | 3.0034 | 1.9829 | 0.4960 | 2.3325 | 0.0673 | 0.1221 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0004 | 0.0000 |
| g-10 | 2.9914 | 1.9991 | 0.5518 | 2.2561 | 0.0673 | 0.1240 | 0.0000 | 0.0054 | 0.0008 | 0.0021 | 0.0000 | 0.0020 | 0.0045 |
or number of cations
[7]:
g.cations(ncat=8, tocat=True)
[7]:
| Si{4+} | Al{3+} | Mg{2+} | Fe{2+} | Mn{2+} | Ca{2+} | K{+} | Na{+} | Ti{4+} | Cr{3+} | Zn{2+} | P{5+} | Y{3+} | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g-09 | 3.0017 | 1.9817 | 0.4957 | 2.3312 | 0.0673 | 0.1220 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0004 | 0.0000 |
| g-10 | 2.9897 | 1.9979 | 0.5515 | 2.2548 | 0.0673 | 0.1239 | 0.0000 | 0.0054 | 0.0008 | 0.0021 | 0.0000 | 0.0020 | 0.0045 |
Cations could be also calculated according to mineral structural formula using Mineral instance.
[8]:
grt = mindb.Garnet()
apfu = g.apfu(grt)
apfu
[8]:
| Si{4+} | Al{3+} | Mg{2+} | Fe{2+} | Mn{2+} | Ca{2+} | K{+} | Na{+} | Ti{4+} | Cr{3+} | Zn{2+} | P{5+} | Y{3+} | Fe{3+} | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g-09 | 3.0017 | 1.9817 | 0.4957 | 2.3175 | 0.0673 | 0.1220 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0004 | 0.0000 | 0.0137 |
| g-10 | 2.9897 | 1.9979 | 0.5515 | 2.2409 | 0.0673 | 0.1239 | 0.0000 | 0.0054 | 0.0008 | 0.0021 | 0.0000 | 0.0020 | 0.0045 | 0.0139 |
Cations calculated according to mineral structural formula, could be used to calculate endmembers proportios (if defined for given mineral).
[9]:
apfu.endmembers()
[9]:
| Alm | Prp | Sps | Grs | Adr | Uv | CaTi | |
|---|---|---|---|---|---|---|---|
| g-09 | 0.771849 | 0.165090 | 0.022414 | 0.040367 | 0.000280 | 0.000000 | 0.000000 |
| g-10 | 0.751060 | 0.184849 | 0.022558 | 0.041276 | 0.000197 | 0.000044 | 0.000016 |
Recalculate plagioclase…
[10]:
f = d.search("pl-")
f
[10]:
| SiO2 | Al2O3 | MgO | FeO | MnO | CaO | K2O | Na2O | TiO2 | Cr2O3 | ZnO | P2O5 | Y2O3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pl-08 | 61.839 | 24.661 | 0.000 | 0.484 | 0.015 | 5.738 | 0.032 | 7.917 | 0.000 | 0.010 | 0.019 | 0.025 | 0.000 |
| pl-22 | 60.657 | 25.087 | 0.001 | 0.000 | 0.000 | 5.849 | 0.106 | 7.940 | 0.000 | 0.000 | 0.003 | 0.113 | 0.000 |
| pl-23 | 61.338 | 25.025 | 0.015 | 0.030 | 0.003 | 5.801 | 0.079 | 7.997 | 0.047 | 0.000 | 0.057 | 0.064 | 0.000 |
[11]:
plg = mindb.Feldspar()
apfu = f.apfu(plg)
apfu
[11]:
| Si{4+} | Al{3+} | Mg{2+} | Fe{2+} | Mn{2+} | Ca{2+} | K{+} | Na{+} | Ti{4+} | Cr{3+} | Zn{2+} | P{5+} | Y{3+} | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pl-08 | 2.7240 | 1.2803 | 0.0000 | 0.0178 | 0.0006 | 0.2708 | 0.0018 | 0.6761 | 0.0000 | 0.0003 | 0.0006 | 0.0009 | 0.0000 |
| pl-22 | 2.6968 | 1.3145 | 0.0001 | 0.0000 | 0.0000 | 0.2786 | 0.0060 | 0.6844 | 0.0000 | 0.0000 | 0.0001 | 0.0043 | 0.0000 |
| pl-23 | 2.7076 | 1.3019 | 0.0010 | 0.0011 | 0.0001 | 0.2744 | 0.0044 | 0.6844 | 0.0016 | 0.0000 | 0.0019 | 0.0024 | 0.0000 |
[12]:
apfu.endmembers()
[12]:
| An | Ab | Or | |
|---|---|---|---|
| pl-08 | 0.285439 | 0.712666 | 0.001895 |
| pl-22 | 0.287517 | 0.706279 | 0.006204 |
| pl-23 | 0.284836 | 0.710546 | 0.004619 |
You can also find out cations p.f.u. not only from your analysis, but also from site occupancies for given mineral structural formula.
[13]:
apfu.mineral_apfu()
[13]:
| Si{4+} | Al{3+} | Mg{2+} | Fe{2+} | Mn{2+} | Ca{2+} | K{+} | Na{+} | Ti{4+} | Cr{3+} | Zn{2+} | P{5+} | Y{3+} | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pl-08 | 2.7240 | 1.2760 | 0.0000 | 0.0000 | 0.0000 | 0.2708 | 0.0018 | 0.6761 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| pl-22 | 2.6968 | 1.3032 | 0.0000 | 0.0000 | 0.0000 | 0.2786 | 0.0060 | 0.6844 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| pl-23 | 2.7076 | 1.2924 | 0.0000 | 0.0000 | 0.0000 | 0.2744 | 0.0044 | 0.6844 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
The difference between analysis and site occupancies could by accessed by property reminder
[14]:
apfu.reminder
[14]:
| Si{4+} | Al{3+} | Mg{2+} | Fe{2+} | Mn{2+} | Ca{2+} | K{+} | Na{+} | Ti{4+} | Cr{3+} | Zn{2+} | P{5+} | Y{3+} | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pl-08 | 0.0 | 0.004246 | 0.000000 | 0.017830 | 0.000560 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000348 | 0.000618 | 0.000932 | 0.0 |
| pl-22 | 0.0 | 0.011310 | 0.000066 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000098 | 0.004253 | 0.0 |
| pl-23 | 0.0 | 0.009496 | 0.000987 | 0.001107 | 0.000112 | 0.0 | 0.0 | 0.0 | 0.001561 | 0.000000 | 0.001858 | 0.002392 | 0.0 |
Plotting
The module plotting providing some common plots, e.g. garnet profiles. Here is quick example
[15]:
gp = Oxides.from_examples("grt_profile")
em = gp.apfu(grt).endmembers()
minplot.plot_grt_profile(em, percents=True)

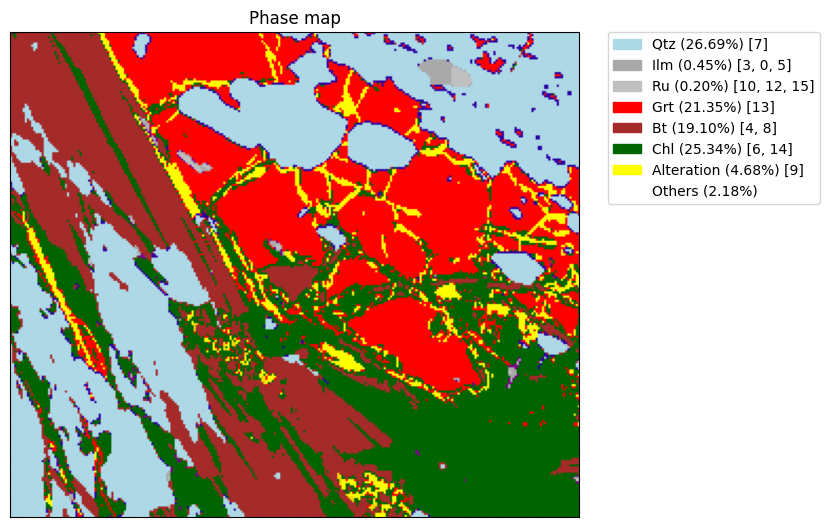
EDS maps
[16]:
from epmatools.maps import MapStore
[17]:
h5 = MapStore.from_examples('ex1')
h5.samples
[17]:
['Demo']
[18]:
s = h5.get_sample('Demo')
[19]:
s.show()

[20]:
s.show('Ca')

[21]:
s.show('Fe/(Fe+Mg)')

[22]:
s.phasemap()
[ ]: